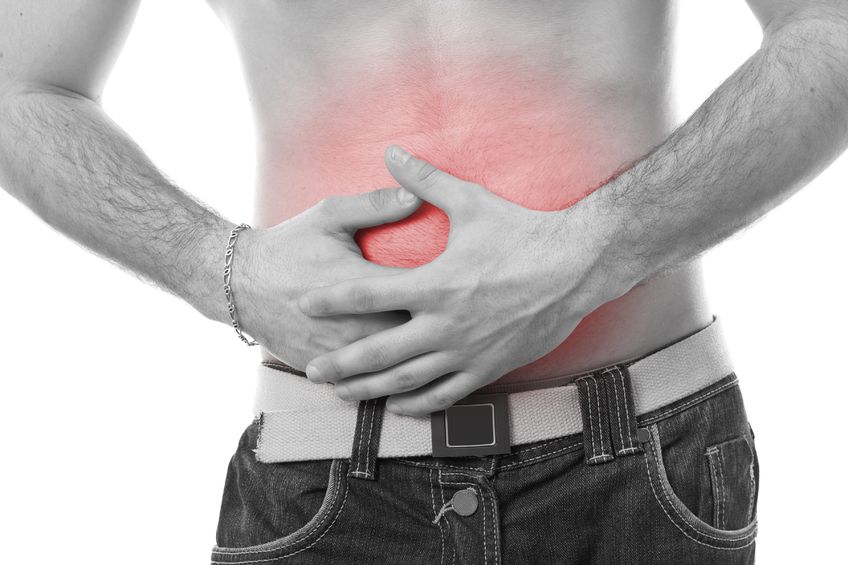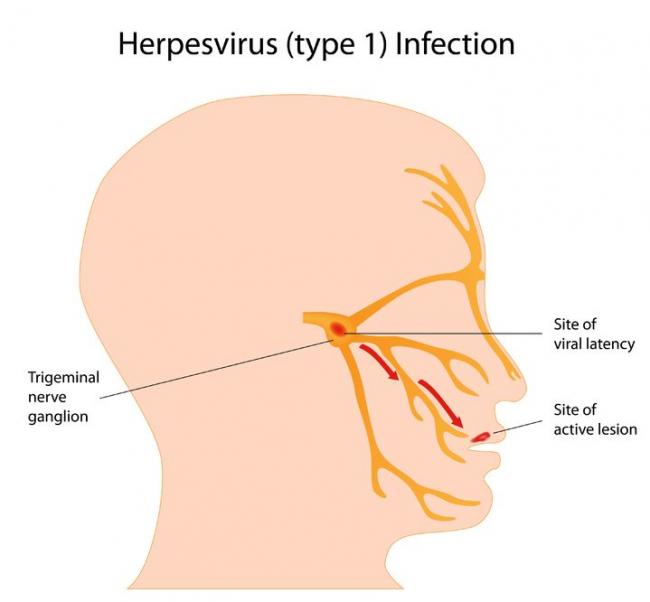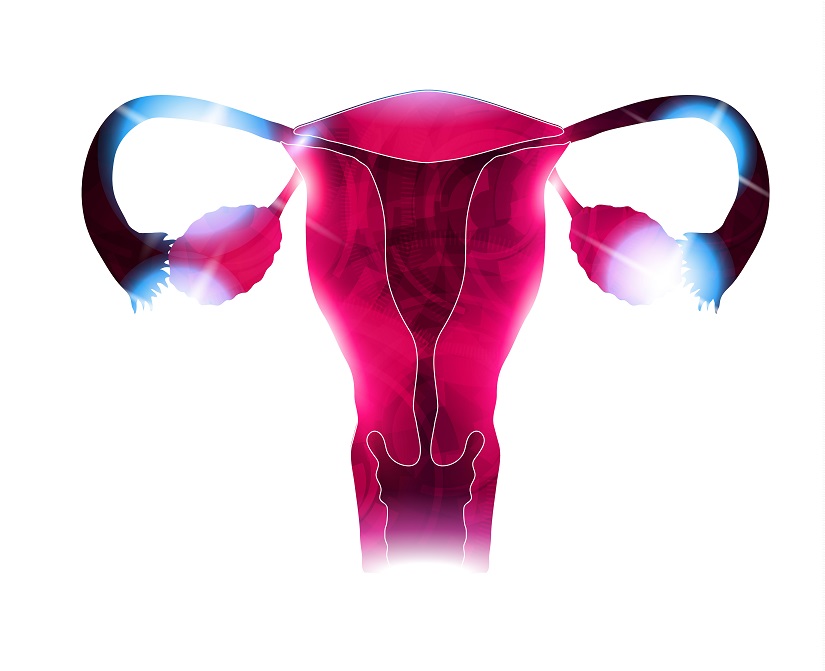


 L’endométriose se caractérise par la présence de tissu endométrial en dehors de l’utérus. Ces lésions tissulaires réagissent à des stimuli hormonaux, comme les œstrogènes, et peuvent causer de très fortes douleurs pelviennes chez bien des femmes. La maladie présente de nombreux stades et degrés différents.
L’endométriose se caractérise par la présence de tissu endométrial en dehors de l’utérus. Ces lésions tissulaires réagissent à des stimuli hormonaux, comme les œstrogènes, et peuvent causer de très fortes douleurs pelviennes chez bien des femmes. La maladie présente de nombreux stades et degrés différents.
 Les infections vaginales amènent régulièrement les femmes soit à consulter leur médecin, soit à se soigner par elles même avec des remèdes en vente libre. La tentation de l’autoprescription surgit vite avec autant de solutions disponibles pour les infections à levures.
Les infections vaginales amènent régulièrement les femmes soit à consulter leur médecin, soit à se soigner par elles même avec des remèdes en vente libre. La tentation de l’autoprescription surgit vite avec autant de solutions disponibles pour les infections à levures. 

 Les autres symptômes sont notamment l’augmentation de la fréquence urinaire, et une sensation d’urgence et de pression dans la région pelvienne. L’objet de cet article est de passer en revue les facteurs-clés contribuant à la CI ainsi que les thérapies basées sur l’expérience permettant de traiter chacun de ces facteurs.
Les autres symptômes sont notamment l’augmentation de la fréquence urinaire, et une sensation d’urgence et de pression dans la région pelvienne. L’objet de cet article est de passer en revue les facteurs-clés contribuant à la CI ainsi que les thérapies basées sur l’expérience permettant de traiter chacun de ces facteurs.
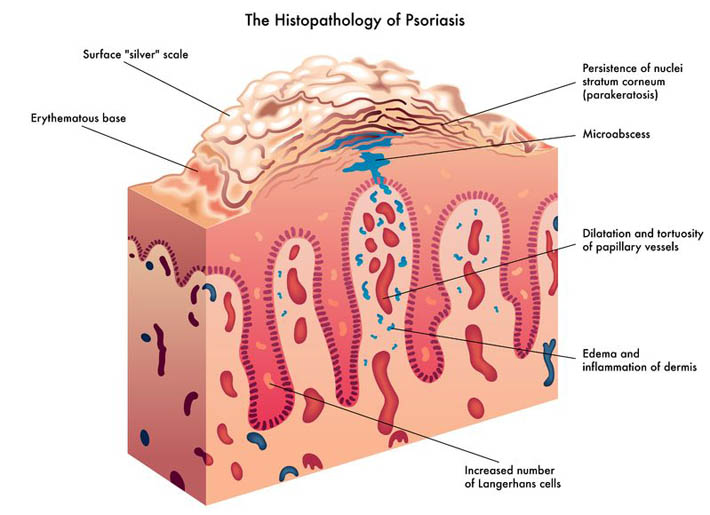
 Le terme dysplasie cervicale se réfère aux cellules anormales présentes sur la surface du col de l’utérus considérées précancéreuses et pouvant mener au cancer. 1) La dysplasie cervicale est principalement causée par une infection transmise sexuellement aux différentes souches du virus du papillome humain (VPH). Des souches différentes peuvent être impliquées autant dans les lésions bénignes que malignes, ce qui signifie que la progression de la maladie semble être dépendante de facteurs individuels. Les études suggèrent qu’une exposition au VPH est l’événement initiateur pouvant entrainer le développement de la dysplasie cervicale, aussi nommée néoplasie intraépithéliale cervicale (CIN).
Le terme dysplasie cervicale se réfère aux cellules anormales présentes sur la surface du col de l’utérus considérées précancéreuses et pouvant mener au cancer. 1) La dysplasie cervicale est principalement causée par une infection transmise sexuellement aux différentes souches du virus du papillome humain (VPH). Des souches différentes peuvent être impliquées autant dans les lésions bénignes que malignes, ce qui signifie que la progression de la maladie semble être dépendante de facteurs individuels. Les études suggèrent qu’une exposition au VPH est l’événement initiateur pouvant entrainer le développement de la dysplasie cervicale, aussi nommée néoplasie intraépithéliale cervicale (CIN).